Резюме
Приведен краткий обзор данных литературы об этиологии, патогенезе, распространенности и клинических проявлениях редко встречающегося врожденного системного заболевания скелета, известного под названием «болезнь Камурати — Энгельмана». Синонимы заболевания: «врожденный системный гиперостоз», «прогрессирующая диафизарная дисплазия», «генерализованный гиперостоз». В основе заболевания лежат нарушения или изменения (мутации) в гене TGFB1, в составе хромосомы 19 (19q13.1). Поражение костей характеризуется медленным прогрессированием и отличается полиморфизмом — от бессимптомных до ярко выраженных форм с болевым синдромом, деформациями сегментов конечностей и контрактурами суставов. Для данного заболевания характерно отставание в физическом развитии, дети плохо прибавляют в весе, отмечается гипогонадизм. Лечение заболевания в основном симптоматическое — направлено на уменьшение болевого синдрома, коррекцию имеющихся деформаций. Прогноз для жизни благоприятный, в отношении функции нижних конечностей — зависит от тяжести и течения заболевания.
Представлено клиническое наблюдение заболевания Камурати — Энгельмана у мальчика в возрасте 13 лет, жителя Закарпатской области. Жалобы на постоянную общую слабость, быструю утомляемость, сонливость, плохой аппетит, деформацию обеих голеней и бедер, боли в голенях при ходьбе. Первое проявление заболевания — деформация голеней отмечена в возрасте 4–5 лет. Общее состояние ребенка удовлетворительное. Телосложение астеническое, пропорциональное, пониженного питания. Мышцы туловища и конечностей развиты слабо. Осанка сутулая. Отмечаются умеренная антекурвация и варусная деформация бедер в проксимальном отделе, выраженная антекурвация голеней. Походка аритмичная, с широкой постановкой стоп. Приседает и встает с большим трудом из-за слабости и боли в области голеней. На рентгенограммах бедренных костей — варусная деформация и антекурвация на уровне диафизов, неравномерный гиперостоз, преимущественно задней, латеральной и медиальной стенок. На рентгенограммах костей обеих голеней — искривление, преимущественно большеберцовых костей под углом, открытым кзади. Неравномерный гиперостоз в области диафизов обеих костей голеней, преимущественно в области задней стенки и проксимальном отделе диафизов, в области передней стенки большеберцовых костей, неравномерное изменение поперечного размера костномозгового канала. Результаты электрохемилюминесцентного исследования маркеров костного метаболизма свидетельствуют о его нарушении, очевидно, вследствие повышенной продукции остеобластами остеокальцина
Наведено короткий огляд даних літератури про етіологію, патогенез, поширеність та клінічні прояви вродженого системного захворювання скелета, що рідко спостерігається, відомого під назвою «хвороба Камураті — Енгельмана». Синоніми захворювання: «вроджений системний гіперостоз», «прогресуюча діафізарна дисплазія», «генералізований гіперостоз». В основі захворювання лежать порушення або зміни (мутації) у гені TGFB1, у складі хромосоми 19 (19q13.1). Ураження кісток характеризується повільним прогресуванням та відрізняється поліморфізмом — від безсимптомних до виражених форм із больовим синдромом, деформаціями сегментів кінцівок та контрактурами суглобів. Для даного захворювання характерним є відставання у фізичному розвитку, діти погано набирають вагу, відмічається гіпогонадизм. Лікування переважно симптоматичне — спрямовано на зменшення больового синдрому, корекцію наявних деформацій. Прогноз для життя сприятливий.
Представлено клінічне спостереження захворювання Камураті — Енгельмана в хлопчика 13 років, мешканця Закарпатської області. Скарги на постійну загальну слабкість, швидку втому, сонливість, поганий апетит, деформацію обох гомілок та стегон, болі в гомілках під час ходи. Перші ознаки захворювання — деформацію гомілок відмічено у віці 4–5 років. Загальний стан дитини задовільний. Статура астенічна, пропорціональна, зниженого харчування. М’язи тулуба і кінцівок слабо розвинуті. Постава сутула. Відмічаються помірна антекурвація та варусна деформація стегон у проксимальному відділі, значна антекурвація гомілок. Хода аритмічна, із широкою постановкою стоп. Присідає і встає з великими труднощами через болі в гомілках. На рентгенограмах стегнових кісток — варусна деформація та антекурвація на рівні діалізів, нерівномірний гіперостоз, переважно задньої, медіальної та латеральної стінок. На рентгенограмах кісток гомілок — скривлення, переважно великогомілкових кісток під кутом, відкритим дозаду. Нерівномірний гіперостоз у ділянці діалізів обох кісток гомілок, переважно в ділянці задньої стінки та передньої стінки проксимального відділу діалізу, нерівномірні зміни поперечного розміру кістково-мозкового каналу. Результати електрохемілюмінесцентного дослідження маркерів кісткового метаболізму свідчать про його порушення, очевидно, внаслідок підвищеної продукції остеобластами остеокальцину.
A brief review of the literature on the etiology, pathogenesis, prevalence and clinical manifestations of rare congenital systemic disease of the skeleton, known as Camurati-Engelmann disease, is presented. Synonyms of the disease: congenital systemic hyperostosis, progressive diaphyseal dysplasia, generalized hyperostosis. Disorders or changes (mutations) in TGFB1 gene, as a part of chromosome 19 (19q13.1), underlie the disease. Bone lesions are characterized by a slow progression and polymorphism — from asymptomatic to distinct forms with pain syndrome, deformities of limb segments and joint contractures. This disease is characterized by lag in physical development, children gain weight poorly, there is hypogonadism. Treatment of the disease is mainly symptomatic — it is aimed at reduction of pain syndrome, correction of existing deformities. Prognosis is favorable, in terms of the lower extremities function — depends on the severity and course of the disease.
There is presented a clinical observation of Camurati-Engelmann disease in a boy aged 13 years, a resident of the Transcarpathian region. There are complaints about the permanent general weakness, fatigue, drowsiness, poor appetite, deformation of both lower legs and hips, pain in the legs when walking. The first manifestation of the disease — lower leg deformity — is detected at the age of 4–5 years. The general condition of the child is satisfactory. Asthenic body build, proportional, undernourished. The muscles of the trunk and limbs are underdeveloped. The posture is stooped. There is a modest antecurvation and varus deformity in the proximal femur, significant antecurvation of the lower legs. The gait is irregular, with a broad statement of the feet. The patient squats and gets up with great difficulty because of weakness and pain in the shins. On the femoral X-ray — varus deformity and antecurvation at diaphysis, irregular hyperostosis, mainly in the rear, lateral and medial walls. On the radiographs of both shin bones — curvature, mainly of tibia, angle wise, open posteriorly. Irregular hyperostosis in the diaphysis of both shinbones, especially in the area of the rear wall and the proximal diaphysis, in the anterior wall of the tibia, the non-uniform changes in the transverse dimension of the medullary canal. The findings of electrochemiluminescence study of the markers of bone metabolism indicate its disorder, apparently due to increased production of osteocalcin by osteoblasts.
Статья опубликована на с. 103-107
Итальянский ортопед М. Camurati в 1922 г. и немецкий хирург и ортопед G. Engelmann в 1929 г. описали заболевание, которое проявлялось в детском возрасте болями в костях конечностей, прогрессирующей мышечной усталостью и атрофией мышц, нарушением походки («вразвалочку»), симметричным утолщением кортикального слоя длинных трубчатых костей. В литературе заболевание приводится как болезнь Камурати — Энгельмана (БКЭ). Синонимы заболевания: «врожденный системный гиперостоз», «прогрессирующая диафизарная дисплазия», «генерализованный гиперостоз» [2, 4, 8].
БКЭ — редко встречающееся системное врожденное заболевание скелета. Согласно информации из интернет-порталов ORPHANET (The portal for rare diseases and orphan drugs) и National Organisation for rare disorders (NORD), до 2013 г. было описано около 300 случаев этого заболевания [5, 8].
В основе заболевания лежат нарушения или изменения (мутации) в гене TGFB1, в составе хромосомы 19 (19q13.1). Ген TGFB1 содержит информацию для синтеза белка, называемого трансформирующим фактором роста бета-1 (TGF-beta-1). Белок TGF-beta-1 помогает контролировать рост и пролиферацию клеток, процесс, посредством которого клетки созревают для выполнения конкретных функций (дифференциация), движение (подвижность) и саморазрушение клеток (апоптоз). Заболевание наследуется по аутосомно-доминантному типу [3].
Болезнь проявляется не сразу после рождения ребенка, а значительно позднее. Дети сравнительно поздно становятся на ножки — на третьем — четвертом году жизни, при этом иногда испытывают затруднения при ходьбе. Во многих случаях наблюдается отставание в физическом развитии, дети растут худыми, бледными, плохо прибавляют в весе, отмечается гипогонадизм [5, 6]. Нередко отмечаются некоторая общая и мышечная слабость, сонливость. В остальном же больные ничем не отличаются от своих сверстников, имеют нормальный внешний облик.
Поражение костей характеризуется медленным прогрессированием и отличается полиморфизмом — от бессимптомных до ярко выраженных форм с болевым синдромом, деформациями сегментов конечностей и контрактурами суставов. Боли в костях отмечаются в 94 % случаев и описываются пациентами как постоянные, ноющие. Характерно усиление боли в связи с двигательной активностью, стрессом или охлаждением [5, 8]. У детей младшего возраста гиперостоз в основном носит несимметричный, очаговый характер. Участки с утолщением кортикального слоя чередуются с участками, где эти изменения отсутствуют. С возрастом гиперостоз становится симметричным и приобретает генерализованный характер. В 52 % случаев утолщение диафиза определяется при пальпации. В ряде случаев наблюдается цилиндрическая форма пораженных сегментов. Могут наблюдаться сгибательные контрактуры в тазобедренных и коленных суставах. Проявлением заболевания могут быть сглаженность поясничного лордоза, кифоз, сколиоз, плоскостопие, coxa valga и genu valga [4, 5, 8]. Вероятность возникновения переломов в области поражения снижается в связи с увеличением минеральной плотности кости. Однако при этом возможна замедленная консолидация переломов [8]. В литературе описаны в основном спорадические случаи заболевания. Оно встречается в различных этнических группах, не зависит от пола. Частота и распространенность заболевания не установлены, так как нередко оно протекает бессимптомно и выявляется случайно при рентгенологическом исследовании [4]. Y. Makita et al. (2000) описали различные рентгенологические проявления БКЭ у 12 пациентов в 3 поколениях одной японской семьи [7].
Первое проявление БКЭ возможно в любом возрасте. По данным портала GeneReviews® [http://www.ncbi.nlm.nih.gov/books/NBK1156/] (последнее обновление 05.03.2015), среди зарегистрированных 306 пациентов в возрасте от 1 года до 76 лет средний возраст, в котором обнаружены симптомы заболевания, составил 13,4 года [8, 9].
Диагностика заболевания основана на результатах рентгенологического исследования пораженных отделов опорно-двигательного аппарата. Анализы крови не показывают характерных морфологических и биохимических изменений, в частности не характерна анемия [2, 4, 9].
При изучении биохимических маркеров костного метаболизма установлены возможные аспекты патогенеза нарушений костного ремоделирования вследствие индукции синтеза соматотропного гормона и паратгормона. В результате комплексного разнонаправленного влияния данных гормонов происходит разобщение процессов синтеза и резорбции костной ткани на фоне общего замедления костного ремоделирования [1]. Лечение БКЭ в основном симптоматическое — направлено на уменьшение болевого синдрома, коррекцию имеющихся деформаций. При выраженном болевом синдроме возможно применение глюкокортикоидных препаратов. Прогноз для жизни благоприятный, в отношении функции нижних конечностей — зависит от тяжести и течения заболевания [4, 5, 7, 8].
Приводим собственное клиническое наблюдение.
Ребенок Р., 13 лет. Жалобы на постоянную общую слабость, быструю утомляемость, сонливость, плохой аппетит, деформацию обеих голеней и бедер, боли в голенях при ходьбе. Мальчик — второй ребенок в семье. Семья постоянно проживает в Закарпатской области Украины. У родителей и старшего брата подобных клинических признаков заболевания не выявлено.
Ребенок с раннего детства отстает в физическом развитии от сверстников. Стоять начал в возрасте 1 г. 4 мес. В возрасте 4–5 лет родители отметили у ребенка легкую хромоту и незначительную деформацию левой, а затем и правой голени — дугообразное искривление кпереди на уровне диафиза. На рентгенограммах отмечена саблевидная деформация большеберцовых костей, гиперостоз задней стенки. Постепенное прогрессирование деформации голеней сопровождалось увеличением их объема. Появилась быстрая утомляемость мышц ног и боли в мышцах голеней при ходьбе. В январе 2015 г. в результате незначительного ушиба наступил косой перелом большеберцовой кости левой голени на уровне нижней трети диафиза с незначительным смещением фрагментов. Лечение проводилось методом иммобилизации гипсовой повязкой. Опороспособность конечности восстановлена через 7–8 недель. Спустя 1 месяц, в апреле 2015 г., во время прыжка с опорой на правую ногу получил закрытый перелом обеих костей правой голени с незначительным смещением — большеберцовой кости в средней трети диафиза, двойной перелом малоберцовой кости на уровне нижней трети и на границе средней и верхней трети (рис. 1). Лечение — иммобилизация гипсовой повязкой. Консолидация — в течение 2–2,5 месяца.
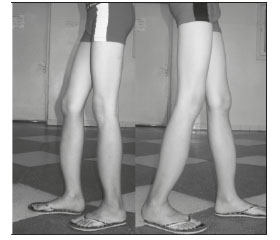
Общее состояние ребенка удовлетворительное. В учебе в школе от сверстников не отстает. Телосложение астеническое, пропорциональное, пониженного питания. Вес тела 38 кг при росте 157 см. Мышцы туловища и конечностей развиты слабо. Осанка сутулая. Незначительное отклонение оси позвоночника вправо в нижнегрудном отделе (рис. 2).
Движения в позвоночнике не ограниченны, безболезненны. Движения в суставах рук без особенностей. Отмечаются умеренная антекурвация и варусная деформация бедер в проксимальном отделе, выраженная антекурвация голеней. Неравномерное увеличение переднезаднего размера голеней на разных уровнях. Умеренно болезненны пассивные и активные движения, а также пальпация в области тазобедренных, коленных и голеностопных суставов. Объем движений в них не ограничен. Походка аритмичная, с широкой постановкой стоп. Приседает и встает с большим трудом из-за слабости и боли в области голеней.
На рентгенограммах бедренных костей — варусная деформация и антекурвация на уровне диафизов, неравномерный гиперостоз, преимущественно задней, латеральной и медиальной стенок (рис. 3).
На рентгенограммах костей обеих голеней — искривление, преимущественно большеберцовых костей под углом, открытым кзади. Неравномерный гиперостоз в области диафизов обеих костей голеней, преимущественно в области задней стенки и проксимальном отделе диафизов, в области передней стенки большеберцовых костей, неравномерное изменение поперечного размера костномозгового канала (рис. 4).
Результаты электрохемилюминесцентного исследования маркеров костного метаболизма (ГУ «Институт травматологии и ортопедии НАМН Украины») свидетельствуют о его нарушении, очевидно, вследствие повышенной продукции остеобластами остеокальцина (табл. 1). Показатели нормы в табл. 1 приведены в соответствии с результатами исследований Т.А. Галятиной и соавт. (2014) [1].
На основании жалоб, анамнеза заболевания, оценки имеющихся отклонений в общем соматическом состоянии ребенка, а также данных рентгенологического и лабораторного исследований поставлен диагноз: прогрессирующая диафизарная дисплазия (болезнь Камурати — Энгельмана).
В настоящее время ребенок продолжает учебу в школе. Проводится комплексное общеукрепляющее лечение. Ребенок готовится к хирургическому вмешательству с целью коррекции осевой деформации обеих голеней.
Список литературы
1. Галятина Т.А. Особенности регуляции костного ремоделирования при врожденной патологии опорно-двигательного аппарата у детей / Т.А. Галятина, И.М. Устьянцева, О.И. Хохлова // Клиническая лабораторная диагностика. — 2014. — № 4. — С. 17-21.
2. Лагунова И.Г. Клинико-рентгенологическая диагностика дисплазий скелета / И.Г. Лагунова. — М.: Медицина, 1989. — 256 с.
3. A family with Camurati-Engelman disease. The role of the missense p.R218C mutation in TGFB1 in bones and endocrine glands / Toumba M., Neocleous V., Shammas C. et al. // J. Pediatr. Endocrinol. Metab. — 2013. — № 26(9–10). — Р. 987-993.
4. Camurati-Engelmann disease in a family from Croatian Island: an old bone scan confirmed pattern of inheritance / Baretić M., Korsić M., Potocki K. et al. // Coll. Antropol. — 2014. — № 38(2), Jun. — P. 755-758.
5. Camurati-Engelmann disease: review of the clinical, radiological, and molecular data of 24 families and implications for diagnosis and treatment / Janssens K., Vanhoenacker F., Bonduelle M. et al. // J. Med. Genet. — 2006. — № 43. — P. 1-11.
6. Gupta S. Camurati-Engelmann disease in conjunction with hypogonadism / Gupta S., Cheikh I.E. // Endocrinol. Pract. — 2005. — № 11. — P. 399-407.
7. Intrafamilial phenotypic variability in Engelman disease (ED): are ED and Ribbing disease the same entity? / Makita Y., Nishimura G., Ikegawa S. et al. // Am. J. Med. Ge–net. — 2000. — № 91(2). — Р. 53-56.
8. Marked phenotypic variability in progressive diaphyseal dysplasia (Camurati-Engelmann disease): report of a four-ge–neration pedigree, identification of a mutation in TGFB1, and review/ Wallace S.E., Lachman R.S., Mekikian P.B. et al. // Am. J. Med. Genet. — 2004. — № 129A. — P. 235-247.
9. Skull base manifestations of Camurati-Engelmann di–sease / Carlson M.L., Beatty C.W., Neff B.A. et al. // Arch. Otolaryngol. Head and Neck Surg. — 2010. — № 136. — P. 566-575.

/105.jpg)
/106.jpg)
/106_2.jpg)