Introduction
West syndrome (WS) is characterized by a hipsarrhytmia pattern on electroencephalography (EEG), spasm type seizures, and psychomotor regression triad and it is the most common “catastrophic” childhood epileptic syndrome in infancy [1, 2]. The incidence of WS is estimated at 2–5/10.000 newborns, with a preference for males [3, 4]. Spasms occur in 90 % of patients in the first year, most commonly between 4 and 7 months [1]. WS can be replaced by other resistant epileptic syndromes in most patients [5].
In the beginning, infantile spasms were classified as idiopathic, cryptogenic, and symptomatic based on clinical assessment. West syndrome is classified as idiopathic when no etiological cause is identified, cryptogenic when a possible etiology is suspected but not identified, and symptoma-tic group when a definite etiology is detected. Idiopathic epilepsies, on the other hand, are generally based on a genetic predisposition of unknown etiology and generally good prognosis. [5]. The ILAE Commission on Classification and Terminology substituted genetic, structural, metabolic, and unknown aetiologies for “cryptogenic” and “symptomatic” in 2010 [6].
Although there is no definitive evidence for a standard treatment protocol for West syndrome, the effectiveness of three drugs has so far been proven; corticotropin (ACTH), vigabatrin, and corticosteroid. The first choice is adrenocorticotropic hormone (ACTH) or oral steroids, and the second choice is vigabatrin or other antiepileptic drugs. In cases resistant to these three drugs used as the first choice, pyridoxine phosphate, valproic acid, topiramate, clobazam, zonisamide, levetiracetam, the ketogenic diet can be added to the treatment [7–9].
The purpose of this study is to document demographic characteristics, clinical and laboratory findings, treatment responses, neurodevelopmental outcomes, and risk factors developed during long-term follow-up with the diagnosis of WS.
Materials and methods
Patients who were diagnosed with WS between July 2011 and December 2012 were included in the study. Ethical approval has been obtained for the use of file information. The following data were collected from each patient: ages, gender, findings associated with spasms, seizure frequency, duration of follow-up, prenatal, natal, and postnatal stories, family history. Systemic and neurological examinations, cerebral imaging (brain magnetic resonance imaging (MRI) and/or computed tomography (CT) and laboratory findings, and EEG were reviewed. Biochemical tests (uric acid, creatine kinase, blood urea nitrogen, creatinine, thyroid hormones, serum ammonia, lactate) were performed from laboratory tests.
The development of each child was assessed using the Denver 2 developmental screening test, the Ankara Developmental Screening Inventory test. Following the onset of spasms, follow-up types, neurological examination, developmental tests, applied treatments, and side effects and neurological sequelae were recorded. Age at onset of spasms are classified: early-onset: < 3 months (adjusted age for premature), classical onset: 3 months to 12 months, late-onset: 12 months and later.
Etiological classification
The patients are divided into two groups according to a definable cause: the symptomatic group and the idiopathic group. The patient's head circumference measurements were assessed according to age and gender. The time between the start of treatment and the delay of treatment was recorded. The time between the first observation of the spasms and the beginning of the treatment was considered as early if it is lower than 2 months and as late if it is more than 2 months.
Treatments. Adrenocorticotropic hormone (ACTH), vigabatrin (VGB), and corticosteroids are the first-line treatments for WS. Synthetic ACTH preparation (synacthen depot® ampoule) was administered 0.5 mg twice a week for younger than 12 months or under 10 kg; 1 mg twice a week for older than 12 months or more than 10 kg, intramuscularly for 12 weeks. Vigabatrin (75–100 mg/kg/day) was started for those who were unable to respond and who had tuberous sclerosis in the etiology. When side effects of ACTH treatment resulted in hypertension, infection, hyperglycemia, ACTH was discontinued and continued treatment with clonazepam. In cases of resistant hypertension, frequent recurrent infections, ACTH therapy was discontinued and vigabatrin therapy was started. Treatment response was evaluated by clinical and EEG findings.
Clinical response. Spasms were defined as disappearance within 14 days of initiation of treatment and not to repeat spasms at least 28 days after the last spasm.
EEG response. The EEG took approximately 4 weeks after treatment; was defined as complete disappearance of hypsarrhythmia, followed by any spasm period seen as a cluster after the recovery of the spasms, and observation of vague spasms accompanied by EEG changes. The prognosis of the patients was evaluated according to their seizure, motor, and cognitive/neuromotor developmental status after at least 12 months of follow-up. The absence of seizures du-ring the last 6 months, the ability to have motor skills, and developmental characteristics consistent with the patient’s age, was evaluated as a good prognosis.
Statistical analysis
SPSS for Windows 21 package program was used for statistical analysis of the data. Mean ± standard deviation for data with appropriate distribution in the study and median (minimum-maximum) for data with inadequate distribution were specified. The chi-square test was used for the comparison, the t-test was used for the independent samples, and Pearson correlation analysis was used for the correlations. A p-value of less than 0.05 was considered statistically significant.
Results
Clinical and demographic profile
A total of 69 children were screened. Forty-eight (69.6 %) were male and twenty-one (30.4 %) were female. The mean age of the referral was 8.62 ± 7.20 months (median — 8.0). Fifty-six children (81.6 %) were delivered at term. Eighteen children (26.1 %) were small for gestational age. Table 1 summarizes the features of our cases.
Seizure profile at onset. The average age at seizure onset was 6.8 ± 4.9 months. The mean time between the onset of seizures and the initiation of treatment was 3.90 ± 6.09 months (table 1). West syndrome seizures appeared as a single or cluster. It was observed that 27.5 % (19/69) of the patients presented as single seizures and 72.5 % (50/69) of them cluster seizures. Spasm count was at least 2/day and at most 35/day. 43 patients (62.3 %) had flexor, 19 patients (27.5 %) mixed, and 6 patients (8.7 %) had extensor-type spasms. In most patients, seizures were seen while waking up or awake (table 2).
/10.jpg)
Neuroimaging profile. While symptomatic causes were considered in 60 % of cases before cranial imaging, an increase was observed in the number of symptomatic cases with cranial imaging. Cranial imaging studies were performed at least once in all the patients. 55 (80 %) patients had MRI and others had CT. While pathology was detected in 52 (75 %) of them, imaging was normal in 17 patients (24.6 %). The cranial imaging findings of the patients are shown in table 3.
Mean EEG recovery time was 1.17 ± 1.43 months in the symptomatic group and 2.00 ± 2.88 months in the idiopathic group. There was no significant difference between idiopathic and symptomatic groups in terms of incidence of EEG types (p = 0.308).
Etiology
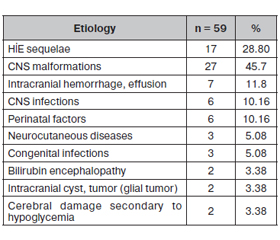
59 (85.51 %) of the patients were in the symptomatic group and 10 (14.49 %) were in the idiopathic group. Congenital CNS malformations (45.7 %) and sequential HIE (28.8 %) were the most frequent causes in the symptomatic group (table 4). There was a significant difference between the idiopathic and symptomatic groups in terms of relapse rates, radiological findings, and prognosis rates (p: 0.035/p < 0.001/p < 0.001). In the idiopathic group, the prognosis was better, relapse rates were low, and radiological findings were less. The prognosis was better in the advanced EEG findings group (p < 0.001). There was no difference between idiopathic and symptomatic groups in terms of seizure type distribution (p: 0.721).
Treatment profile
Before the onset of ACTH therapy, 52 % of patients used antiepileptic drugs, the most frequent was phenobarbital (26.1 %). While 83 % of patients treated with ACTH, treatment was responded, 17 % had resistant seizures. In 64 patients treated with ACTH, side effects developed in various clinical forms. The most common side effect was restlessness. There was no statistically significant difference between the treatment responses with ACTH and other treatments (p = 0.093).
Early response was assessed as partial or complete loss of seizures after ACTH treatment at 1 week. 44 patients (63.8 %) had an early response to ACTH treatment. The prognosis was much better in the patients that started an early treatment (p < 0,001). Relapse was found in 43.5 % of patients after treatment. Clinical response to ACTH treatment was found to be effective regardless of etiology.
Developmental outcomes
55 children were assessed using the Denver 2 developmental screening test, the Ankara Developmental Screening Inventory test [10, 11].
Denver II consists of 134 items evaluating four deve-lopmental areas: personal-social, fine motor-adaptive, language, and gross motor. After calculating the age of the child, it is seen what skills the child should be able to draw by drawing the age line, the tester evaluates the compliance of the child’s development with the age.
Ankara Developmental Screening Inventory test provides an opportunity to identify developmental delay and to reco-gnize the developmentally at-risk babies and children at an early stage and take early interventions. It consists of 154 questions asked to primary caregivers about the development of children. For the other patients for whom these tests could not be performed, clinical observations and family information were used. In our study, 46 (83.6 %) were found to be a developmental delay.
Discussion
A comprehensive clinical evaluation, EEG evaluation, cranial MRI, and genetic and metabolic tests aid early diagnosis. In 277 incidence series of Lombroso [12], the incidence of WS cases with an onset age of 12 months or more was reported as 10 %. Consistent with the literature, the rate of spasms that started after 12 months in our study was 12.5 % and the age of onset of WS was between 3 months and 12 months in 67.9 % of the patients. In our study, patients applied to a health center after an average of 3.9 months after the spasms started. In developed countries, the time from the onset of spasms to diagnosis is reported between 1 and 1.5 months [13–15], while in developing countries this time can be extended to 7.9 months [16]. In our study the percentage of males was 69.56 % and the male: female ratio was 2.28 : 1.
As reported in our literature, WS is reported to be more frequent in males [5, 16, 17].
In WS, the difference between the symptomatic and idiopathic groups is of practical importance. This classification helps to predict prognosis. Patients in the idiopathic group respond very well to treatment and their prognosis is much better. In our study, the symptomatic group rate was higher (85.51 %). Congenital CNS malformations (45.7 %) and sequential HIE/perinatal asphyxia (28.8 %) were the most frequent causes in our study. While perinatal asphyxia is the most common cause in developing countries, prenatal causes such as cortical malformation, neurocutaneous diseases, genetic-metabolic diseases occur in developed countries. The most common risk factor is perinatal asphyxia in our study. In Gupta et al study, asphyxia and other adverse perinatal events are the most common causes [18]. Perhaps this will change with the improvement of prenatal services, institutional distribution, guidance system, and perinatal care [19].
Seizures may be hardly noticeable in the form of a slight shake, or in the form of a tonic contraction that can last for 10 seconds following a rapid spike of 1–2 seconds [2]. Sometimes there is no tonic phase seen afterward. As the number of seizures can be 1–2 per day, the number of seizures can be up to 100. The duration of seizure clusters may vary from 1 minu-te to 10 minutes. In our cases, flexor spasm was observed in 62 % of the patients, extensor spasms were seen in 8.7 % and mixed type spasms were observed in 27.5 % of the patients. In the study of Yilmaz et al. [17] studied 79 % of patients with flexor spasms that is similar to our study.
One of the diagnostic parameters of WS is hypsarrhythmia in the EEG. In our study, there was 43.6 % classic hypsarrhythmia and 11.6 % modified hypsarrhythmia in EEG. EEG findings were consistent with the literature [20]. We observed hypsarrhythmia or modified hypsarrhythmia in EEG (respectively 81.3, 83.3 %) in the symptomatic group. In some idiopathic cases in the literature, the first EEG may be normal or borderline abnormal. In this case, the EEG is required to be withdrawn after 7–10 days [1]. The higher incidence of EEG pathology in the symptomatic group may be associated with the late manifestation of EEG findings in idiopathic cases. We observed 40 % of other seizure types in our study. The most common was myoclonic seizures of the newborn period with 20.3 %. The most common abnormal neurological examination findings were tonus changes, mental retardation, increased deep tendon reflexes, and spasticity. The cerebral palsy rate was 47.3 % at admission. The literature has also been reported between 44 and 67 % [17, 18].
Imaging studies have significantly contributed to the etiology of WS. Abnormal imaging findings are associated with 80 % of WS patients. In literature, cerebral atrophy is the most commonly identified, whereas our study rate was 74.6 % similar to the literature. As is known, the prognosis in the symptomatic group is poor. Structural cerebral anomalies have been reported to significantly affect the prognosis adversely [21]. It is not surprising that the prognosis is poor in this group since the presence of abnormal neurological findings indicates that the cases are symptomatic. Tuberous sclerosis and epilepsy are frequently encountered in studies related to etiology [22]. In our study, tuberous sclerosis was seen in 3.89 %.
Seizures in WS are generally resistant to antiepileptic drugs. The most important cause of the difficulties in treatment is the differences in etiology. ACTH is the best medication proven in treatment. Vigabatrin is preferred in cases with tuberous sclerosis and in cases where ACTH is unresponsive [8]. Lux et al. received treatment response in 76 % of patients receiving ACTH [2]. Hrachovy et al. reported that 75 % of patients using ACTH received a response within the first 2 weeks and relapsed in 31 % of long-term follow-up [23]. In our study, the response rate was 83 % and the relapse rate was 40 %. 63.8 % of the patients had an early response to ACTH treatment. In our study, we found that there was no significant difference in response to ACTH and vigabatrin treatment modalities (p > 0.05) as in the study of Ibrahim et al. [24].
Idiopathic and symptomatic groups had similar responses to ACTH treatment. In both groups, spasms were diminished or disappeared at similar rates. In our study, the rate of response to treatment was high at the early onset of ACTH treatment. Partikian and Mitchell reported that 159 WS patients who were followed for at least one year were seen resistant epilepsy in 53.2 % [25]. In our study, it was 49.2 % were seen as resistant to epilepsy. Koo et al. found that the rates of conversion to neurological sequelae and other epilepsy in the cryptogenic group were lower than in the symptomatic group [26]. In our study, the symptomatic group prognosis (resistant epilepsy recurrence, psychomotor retardation, neurological deficit) and relapse rates were significantly higher than in the idiopathic group (p < 0.001/p = 0.035/p < 0.001). The epileptic prognosis was worse in relapsed patients after treatment (p = 0.029). The treatment response did not have any effect on relapse and epileptic transformation.
Hrachovy and Glaze [27] reported that spasms were accompanied by autonomic and non-spasm findings at varying rates. In our study, 42 % of the patients had abnormal eye movements and 34.8 % of the patients had pre-seizure tremors; were the most common accompanying symptoms.
46 of 55 children (83.6 %) had developmental delay. Consistent with our study, Gupta et al. found that 90.1 % of children had a developmental delay [18]. In the study of Yılmaz et al. With 269 patients, the deve-lopmental prognosis of the cases was assessed by Ankara Developmental Screening Inventory; It was shown that 40 (19 %) patients had borderline, 91 patients (42 %) had mild/moderate delay and 57 patients (28 %) had severe development delay [28]. The presence of developmental delay at first admission in WS is a poor prognostic criterion. Developmental delay is more prominent in those with a young onset age of spasm. In our study, intellectual disability was detected in 54.2 % of patients in the symptomatic group in which ACTH treatment was started early and in 74.2 % of those who were late in the treatment. In a study of Nasiri et al., 67 patients with WS was associated with poor neurodevelopmental outcome. Most developmental delays have also been shown to be due to the presence of symptomatic WS and resistant seizures [29]. Singhi and Ray reported 46.7 % cerebral palsy in the 165 WS patients [30]. The incidence of cerebral palsy in our study was 37.2 % in cases with neurological sequelae. This diffe-rence may be since our records are not well maintained, especially in terms of cerebral palsy.
Conclusions
As a result, WS not only causes resistant seizures but also causes severe neurological sequelae affecting the mental, motor, psychological and sensory development of patients. Spasms can cause more damage to the developing central nervous system. Treatment can reduce psychomotor regression. However, there is currently no optimal treatment for WS. Optimal treatment should be done with at least current treatment protocols. For this reason, early diagnosis, appropriate and adequate treatment may be a positive contribution to the prognosis. Early control of spasms has a positive effect on intellectual development.
Received 01.04.2021
Revised 09.04.2021
Accepted 16.04.2021
Список литературы
1. Alonzo R.D., Rigante D., Mencaroni E., Esposito S. West Syndrome: A Review and Guide for Paediatricians. Clin. Drug. Investig. 2018. 38. 113-124.
2. Lux A.L., Osborne J.P. A proposal for case definitions and outcome measures in studies of infantile spasms and West syndrome: consensus statement of the West Delphi group. Epilepsia. 2004. 45. 1416-1428.
3. Wilmshurst J.M., Ibekwe R.C., O’Callaghan F.J.K. Epileptic spasms — 175 years on: trying to teach and old dog new tricks. Seizure. 2017. 44. 81-86.
4. Brna P.M., Gordon K.E., Dooley J.M., Wood E.P. The epidemiology of infantile spasms. Can. J. Neurol. Sci. 2001. 28. 309-312.
5. Pellock J.M., Hrachovy R., Shinnar S. et al. Infantile spasms: a U.S. consensus report. Epilepsia. 2010. 51. 2175-2189.
6. Berg A.T., Berkovic S.F., Brodie M.J., Buchhalter J., Cross J.H., Van Emde Boas W. et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE commission on classification and terminology 2005–2009. Epilepsia. 2010. 51. 676-685.
7. Wirrell E.C. et al. How should children with West syndrome be efficiently and accurately investigated? Results from the national Infantile Spasms Consortium. Epilepsia. 2015. 56(4). 617-625.
8. Fois A. Infantile spasms: review of the literature and personal experience. Ital. J. Pediatr. 2010, Feb 8. 36. 15.
9. Kossoff E.H., Hedderick E.F., Turner Z., Freeman J.M. A case-control evaluation of the ketogenic diet versus ACTH for new-onset infantile spasms. Epilepsia. 2008. 49. 1504-1509.
10. Yalaz K., Epir S. The Denver Developmental Screening Test: normative data for Ankara children. Turk. J. Pediatr. 1983. 25. 245-258.
11. Savaşır I., Sezgin N., Erol N. Ankara Gelişim Tarama Envanteri (Ankara Developmental Screening Inventory). 2nd ed. Ankara: Türk Psikologlar Derneği (Turkish Psychological Association), 1993.
12. Lombroso C.T. A prospective study of infantile spasms: clinical and therapeuthic consideration. Epilepsia. 1983. 24. 135-158.
13. Zhu X., Chen O., Zhang D. et al. A prospective study on the treatment of infantile spasms with first-line topiramate followed by low-dose ACTH. Epilepsy Res. 2011. 93. 149-154.
14. Lagae L., Verhelst H., Ceulemans B. et al. Treatment and long term outcome in West syndrome: the clinical reality. A multicentre follow up study. Seizure. 2010. 19. 159-164.
15. Cohen-Sadan S., Kramer U., Ben-Zeev B. et al. Multicenter long-term follow-up of children with idiopathic West syndrome: ACTH versus vigabatrin. Eur. J. Neurol. 2009. 16. 482-487.
16. Kaushik J.S., Patra B., Sharma S. et al. Clinical spectrum and treatment outcome of West Syndrome in children from Northern India. Seizure. 2013. 22. 617-621.
17. Yılmaz Ü., Özdemir R. West syndrome: Clinical features and short-term prognosis. Journal of Clinical and Experimental Investigations. 2014. 5(1). 86-92.
18. Gupta J., Sharma S., Mukherjee S.B., Jain P., Aneja S. Neuro-developmental and epilepsy outcomes of children with west syndrome: A cross-sectional study from North India. 2020.
19. Osborne J.P., Lux A.L., Edwards S.W. et al. The underlying etiology of infantile spasms (West syndrome): information from the United Kingdom Infantile Spasms Study (UKISS) on contemporary causes and their classification. Epilepsia. 2010. 51. 2168-2174.
20. Shields W.D. Infantile Spasms: Little Seizures, Big Consequences. Epilepsy Curr. 2006. 6. 63-69.
21. Riikonen R. Epidemiological data of West syndrome in Finland. Brain Dev. 2001. 23. 539-541.
22. Chandra P.S., Salamon N., Nguyen S.T. et al: Infantile spasm-associated microencephaly in tuberous sclerosis complex and cortical dysplasia. Neurology. 2007. 68. 438-445.
23. Hrachovy R.A., Frost J.D. Jr. Severe encephalopathic epilepsy infants: infantile spasms (West syndrome). In: Pellock J.M., Bourgeois B.F.D., Dodson W.E., editors. Pediatric epilepsy: diagnosis and therapy. 3rd ed. New York: Demos, 2008. 249-268.
24. Ibrahim S., Gulab S., Ishaque S., Saleem T. Clinical profile and treatment of infantile spasms using vigabatrin and ACTH — a developing country perspective. BMC Pediatrics. 2010. 10. 1.
25. Partikian A., Mitchell W.G. Mayor adverse events associated with treatment of Infantile spasms. J. Child Neurol. 2007. 22. 1360-1366.
26. Koo B., Hwang P.A., Logan W.J. Infantile spasms: outcome and prognostic factors of cryptogenic and symptomatic groups. Neuro-logy. 1993 Nov. 43(11). 2322-2327.
27. Hrachovy R.A., Glaze D.G., Frost J.D. Jr. A retrospective study of spontaneous remission and long-term outcome in patients with infantile spasms. Epilepsia. 1991. 32. 212-214.
28. Yilmaz S., Tekgul H., Serdaroğlu G., Akçay A., Gokben S. Evaluation of ten prognostic factors affecting the outcome of West syndrome. Acta Neurologica Belgica. 2016. 116. 519-527.
29. Nasiri J., Kachuei M., Kermani R., Samaninobandegani Z. Neurodevelopmental outcomes of the West syndrome in pediatric patients: The first report from the Middle-East. Res. Dev. Disabil. 2019. 89. 114-119. doi: 10.1016/j.ridd.2019.03.010.
30. Singhi P., Ray M. Profile of West syndrome in North Indian children. Brain Dev. 2005 Mar. 27(2). 135-140.

/9.jpg)
/10.jpg)
/11.jpg)