Статтю опубліковано на с. 153-158
Вступ
Провідним напрямком наукової та клінічної роботи відділення психоневрології ДУ «Інститут педіатрії, акушерства і гінекології НАМН України» завжди були і залишаються проблеми перинатальної неврології. Серед них однією з пріоритетних є проблема вивчення рідкісних (раритетних), або так званих орфанних (сирітських), захворювань. У 1983 р. уперше був уведений термін «орфанні хвороби», що охоплює захворювання, частота яких не перевищує 5 випадків на 10 тис. осіб. У США орфанним визнають захворювання, на яке хворіє 1 людина з 1500, в Японії — 1 з 2500, у Росії — 10 зі 100 000 [1].
Сьогодні проблема орфанних захворювань в Україні та світі є надзвичайно актуальною. В Україні законодавчо затверджено термін «орфанне захворювання» з 15 квітня 2014 р., коли було ухвалено Закон України № 1213-VII «Про внесення змін до Основ законодавства України про охорону здоров’я щодо забезпечення профілактики та лікування рідкісних (орфанних) захворювань», згідно з яким рідкісне (орфанне) захворювання визначається як таке, що загрожує життю людини, хронічно прогресує, призводить до скорочення тривалості життя громадянина або до його інвалідності, поширеність якого серед населення не частіше ніж 1 : 2000 [2].
Зважаючи на високий рівень поширеності орфанних захворювань нервової системи в України та високу соціальну важливість надання допомоги пацієнтам, які страждають від них, у 2014 р. відділення дитячої психоневрології ДУ «Інститут педіатрії, акушерства і гінекології НАМН України» змінило назву на «відділення психоневрології для дітей із перинатальною патологією та орфанними захворюваннями».
За даними Європейського комітету експертів з рідкісних захворювань (EUCERD), загальна кількість різних орфанних хвороб досягає 8 тисяч. Встановлено, що 80 % рідкісних захворювань обумовлені генетичними причинами [3].
Особливо актуальною проблема рідкісних хвороб є для педіатрії та клінічної генетики дитячого віку, тому що, за даними Європейської організації хворих із рідкісними захворюваннями (EURORDIS), 75 % рідкісних захворювань проявляються у ранньому дитячому віці, в 65 % випадків вони мають тяжкий інвалідизуючий перебіг, а в 50 % — несприятливий прогноз для життя. Крім того, в 35 % випадків вони є причиною смерті дітей на першому році життя. Майже половина хворих дітей із рідкісними спадковими хворобами страждає від затримки нервово-психічного розвитку [4].
У клініці дитячої психоневрології ДУ «ІПАГ НАМН України» діти, які мають затримку психоемоційного та мовленнєвого розвитку або розлад аутистичного спектра (РАС) у поєднанні з генетичною патологією, проходять всебічне неврологічне обстеження, діагностику із застосуванням магнітно-резонансної томографії головного мозку, електроенцефалографії (ЕЕГ) (за потреби — відео-ЕЕГ-моніторингу), біохімічних і молекулярно-генетичних досліджень.
У цій публікації ми звертаємо увагу на синдром Сотоса — рідкісне непрогресуюче генетичне захворювання, що характеризується прискоренням соматичного розвитку (гігантизмом) і затримкою формування мовлення та інтелекту. У літературі, присвяченій даному синдрому, зазначається, що когнітивна недостатність, затримка розвитку мовлення, порушення поведінки є обов’язковими симптомами захворювання [5]. Однак існує лише кілька описаних випадків поєднання синдрому Сотоса з РАС [6]. У статті ми наводимо клінічний випадок поєднання синдрому Сотоса з РАС.
Історична довідка
Синдром Сотоса був уперше описаний в 1964 р. у статті, опублікованій Juan F. Sotos та співавторами у New England Journal of Medicine. Повідомлялося про групу з 5 дітей, у яких спостерігались особливі клінічні ознаки, що поєднувались у синдром: прискорений соматичний розвиток, акромегалоїдні риси обличчя та непрогресуюча розумова відсталість [7]. У перші роки синдром переважно називався «церебральний гігантизм», однак пізніше ця назва була витіснена «синдромом Сотоса».
У 1992 р. Cole and Hughes в огляді, що базувався на спостереженні 79 пацієнтів віком від 1 до 6 років, запропонували такі критерії захворювання: специфічний фенотип обличчя, прискорений соматичний розвиток, прискорений кістковий вік, затримка інтелектуального розвитку [8].
Значний вклад у встановленні критеріїв діагностики синдрому Сотоса внесла група дослідників із Великої Британії та Японії, якими у 2002 р. було виявлено етіологічний фактор захворювання — мутацію гена NSD1. До цього часу діагноз синдрому Сотоса встановлювався лише клінічно. Загалом, починаючи з 1980 р., опубліковано близько 500 випадків синдрому Сотоса [9].
Клінічні ознаки
Синдром Сотоса (синдром церебрального гігантизму) — це рідкісне генетичне, в більшості випадків спорадичне (описані сімейні випадки захворювання з аутосомно-домінантним типом успадкування) захворювання, що характеризується надмірним збільшенням показників соматичного розвитку протягом перших 2–3 років життя, специфічними рисами обличчя та затримкою психомовленнєвого розвитку. Поєднання даних ознак у дитини дозволяє встановити клінічний діагноз синдрому Сотоса [10].
На підставі аналізу більше ніж 500 пацієнтів із верифікованою мутацією гена NSD1 було показано, що поєднання трьох кардинальних ознак захворювання спостерігається принаймні в 90 % носіїв патологічного гена [11].
Показники поширеності серед новонароджених, за даними різних досліджень, знаходяться в діапазоні від 1 : 10 000 до 1 : 50 000 [12].
Діти з синдромом Сотоса зазвичай мають більший зріст, масу тіла (макросомію) та розміри голови (макроцефалію) при народженні, ніж їх однолітки. Через характерні розміри і форму голови синдром Сотоса іноді називають синдромом церебрального гігантизму. Фенотипові ознаки синдрому можуть варіювати у різних пацієнтів і включають непропорційно великий округлий череп (макроцефалію) з випнутим чолом та сплюснутими скронями, великі руки і ноги. Обличчя в період від 1 до 6 років характеризується наявністю високого широкого чола (голова нагадує перегорнуту грушу — доліхоцефалія), можуть бути залисини у лобно-тім’яних ділянках, гіперемія шкіри на вилицях, антимонголоїдний розріз очей, загострене підборіддя, збільшені щелепи, гіпертелоризм (аномально збільшена відстань між очима), гіпофорія (вертикальний страбізм, спрямований донизу), високе «готичне» піднебіння. У деяких дітей може бути незвичний фенотип, наприклад монголоїдний розріз очей або ненормальний ріст волосся (гіпертрихоз). У юнацькому та шкільному віці специфічні риси обличчя стають менш помітними, хоча звертає на себе увагу підборіддя, що випинає. Близько 90 % дітей мають окружність голови більше 2 стандартних відхилень (SD) [5, 10, 11].
Характерною є виражена гіпотонія при народженні, завдяки чому дитина має вигляд «ляльки з ганчір’я», тобто має місце синдром млявої дитини. Можуть тривало зберігатися проблеми з вигодовуванням, ослаблений смоктальний рефлекс, що може вимагати годування дитини через зонд, часті зригування. Близько 70 % дітей із синдромом Сотоса переносять жовтяницю у неонатальний період. Незважаючи на швидкий приріст фізичних показників, захворювання супроводжується затримкою рухового розвитку. Діти з синдромом Сотоса пізніше починають утримувати голову, сидіти та ходити, що відбувається внаслідок низького м’язового тонусу. Також відзначається моторна незграбність, невпевнена хода. Антропометричні показники продовжують значно випереджати вікові норми (наприклад, у 6-місячних дітей розмір голови може бути таким, як у 18-місячної дитини). Синдром Сотоса характеризується прискоренням росту кісток і кісткового віку у 75 % дітей. Ендокринний статус зазвичай залишається не порушеним [5, 10, 13].
Також психологічний розвиток пацієнтів характеризується затримкою мовленнєвого, когнітивного та соціального розвитку. Синдром може супроводжуватися легкою або помірною інтелектуальною недостатністю, РАС, гіперактивністю з дефіцитом уваги, агресивністю, підвищеною дратівливістю, обсесіями та компульсіями, фобіями, істериками та імпульсивною поведінкою. Майже завжди є проблеми з розвитком мовлення. У пацієнтів часто наявні порушення фонації, може бути заїкання, монотонний голос [6, 14, 15].
У дослідженні Sarimski (2003) повідомляється про те, що у дітей із синдромом Сотоса має місце переважно м’яка інтелектуальна недостатність (59 %) порівняно з середніми показниками в популяції [15].
П’ять досліджень описують особливості аутистичної поведінки серед хворих із синдромом Сотоса. Два дослідження повідомляють про наявність ознак РАС у 46 % пацієнтів [17, 18]. Основні ознаки РАС включають в себе: порушення комунікативних функцій та соціальної взаємодії, стереотипну поведінку, обмеженість або одноманітність інтересів. Інші два дослідження описують хворих з «аутистичною поведінкою», хоча вони не описані докладно [19, 20]. Останнє дослідження Finnegan et al. (1994) свідчить про те, що в деяких дітей, яких обстежували з приводу синдрому Сотоса, була подібна поведінка, як у дітей із синдромом Аспергера. Однак це був суб’єктивний висновок автора, що не мав ніякого діагностичного підтвердження [21]. У цілому, хоча «аутистична поведінка» була зареєстрована в невеликій кількості досліджень, стандартизована оцінка з використанням встановлених діагностичних критеріїв не проводилася. У зв’язку з цим дані щодо поширеності РАС серед пацієнтів із синдромом Сотоса натепер відсутні.
У період старшого дитинства поступово зникають відмінності між здоровими дітьми та пацієнтами з синдромом Сотоса. М’язовий тонус поступово підвищується, у деяких випадках відбувається поліпшення розвитку мовлення. Темпи прискореного соматичного росту уповільнюються у період до початку пубертату, тому підлітки та дорослі, які страждають від синдрому Сотоса, відповідають за антропометричними показниками здоровим одноліткам. У багатьох дорослих із синдромом Сотоса діагностичні показники можуть відповідати нормі за інтелектом та фізичним розвитком.
Описані випадки розвитку злоякісних пухлин у дітей із синдромом Сотоса, але жоден вид онкологічних захворювань не є асоційованим із цим захворюванням. Пухлини зустрічаються приблизно в 3 % осіб із синдромом Сотоса і включають крижово-куприкову тератому, нейробластому, пресакральні та ретросакральні гангліонейроми, гострий лімфобластний лейкоз і дрібноклітинний рак легенів [22, 23]. Скринінг на пухлину Вільямса вважається недоцільним. У наш час немає достатньо доказів того, що діти з синдромом Сотоса мають значно більший ризик розвитку онкологічної патології порівняно із загальною популяцією.
Додаткові критерії синдрому можуть бути наявні у більше ніж 15 % пацієнтів. До таких клінічних ознак належать: прискорений кістковий вік; аномалії головного мозку, що виявляються при КТ- або МРТ-обстеженні; проблеми з годуванням у ранньому віці; жовтяниця та гіпотонія у неонатальний період; епілептичні напади; сколіоз; аномалії серця та нирок; прееклампсія у матері в анамнезі; гіпермобільність суглобів; плоскостопість та інші [13].
Етіологія
Етіологія синдрому Сотоса була встановлена у 2002 р., коли вперше було виявлено мутацію гена NSD1, який кодує гістонову метилтрансферазу — фермент, що бере участь у регуляції функцій хроматину. При цій мутації також виникає порушення кодування ядерного рецепторного білка, який бере участь у регуляції нормального росту і розвитку [24]. Було встановлено, що до розвитку синдрому може призводити транслокація t(5; 8) (q35; q 24.1), що викликає гаплонедостатність (неповне домінування) гена NSD1 (ядерний рецептор SET-домену синтезу білка людини). Пізніше було встановлено, що у більше ніж 90 % випадків за розвиток синдрому відповідає внутрішньогенна мутація у NSD1 та мікроделеція в регіоні 5q35. Зокрема, внутрішньогенна мутація була виявлена у 80–85 % хворих серед європейської популяції, тоді як мікроделеція в регіоні 5q35 — лише у 10 % [9]. Отже, верифікація мутації гена NSD1 є вірогідним методом підтвердження діагнозу синдрому Сотоса, оскільки зустрічається лише при цьому захворюванні серед хворих із даним синдромом. Особливо це було підтверджено у хворих японської популяції. Незрозуміло, як зменшення кількості цього білка в процесі розвитку призводить до затримки психічного розвитку, гігантизму та інших патологічних змін, що характерні для синдрому Сотоса.
Близько 95 % випадків синдрому Сотоса виникають спорадично. Більшість із цих випадків є результатом мутацій de novo, пов’язаних із геном NSD1. Було описано кілька сімейних випадків, вивчення яких допомогло дослідникам встановити, що синдром Сотоса може мати аутосомно-домінантний тип спадкування [9].
Доктор Тревор Коул і його колеги з Великобританії при вивченні пацієнтів із синдромом Сотоса і членів їх сімей виявили такі закономірності:
— у 90 % пацієнтів, які чітко відповідали критеріям синдрому Сотоса, було виявлено мутації гена NSD1;
— близько 10 % дітей із фенотипом, що нагадував синдром Сотоса, або з сумнівним діагнозом мали мутації гена NSD1. Майже всі пацієнти цієї групи мали класичний зовнішній вигляд обличчя і велику голову, але мали менший зріст та кістковий вік;
— у пацієнтів, в яких були відсутні фенотипові риси обличчя для синдрому Сотоса, не було виявлено мутації гена NSD1;
— у батьків дітей, які мали мутації гена NSD1, також були фенотипові особливості синдрому Сотоса [9].
Мутація гена NSD1 не виявлена при інших відомих генетичних захворюваннях, що супроводжуються мегалоцефалією, таких як синдром Беквіт — Відемана і синдром Уївер. Але, як вважають дослідники, за наявності як мінімум трьох клінічних ознак синдрому Сотоса проведення молекулярного аналізу не є обов’язковим [5].
Лікування
На сьогодні етіологічного та патогенетичного лікування синдрому Сотоса не розроблено. За необхідності застосовується симптоматичне лікування (протисудомна, дегідратаційна терапія). Рекомендовано застосовувати трудову терапію та реабілітацію для вироблення правильних стереотипних рухів, навичок самообслуговування, уникнути формування шкідливих стереотипних рухів і патологічної постави. Також рекомендована психолого-педагогічна та логопедична корекція.
Прогноз
Синдром Сотоса не вважається загрозливим для життя захворюванням, загальна тривалість життя порівнянна із загальною популяцією. Раннє втручання, психолого-педагогічна корекція та інклюзія дитини у соціум дозволяють значно покращити якість її життя. Однак координаторні порушення можуть зберігатися і в зрілому віці в поєднанні з соматичними чи психічними відхиленнями.
Клінічний випадок
Хлопчик Р., 4 роки, надійшов до відділення дитячої психоневрології ДУ «Інститут педіатрії, акушерства і гінекології НАМН України» зі скаргами матері на наявність у дитини затримки психомовленнєвого розвитку (говорить лише окремі слова, вимовляє нечітко). Спостерігаються порушення поведінки у вигляді гіперактивності, епізодів агресії (може кусатися). Не використовує вказівний жест. З анамнезу відомо, що дитина народилась від 3-ї вагітності (1-ша вагітність завмерла на 12-му тижні; від 2-ї вагітності хлопчик 7 років страждає від виразкового коліту). Вагітність мала перебіг на фоні загрози переривання на 5–6-му тижні, фетоплацентарної недостатності. Пологи в терміні 40 тижнів, ускладнені слабкістю пологової діяльності, навколоплідні води зеленого кольору. Маса тіла при народженні 3900 г, довжина тіла 56 см (+2 SD), закричав слабо. Окружність голови при народженні 36,8 см, окружність грудної клітки 37 см. За шкалою Апгар отримав 7–8 балів. Виписаний з пологового будинку на 3-тю добу.
Розвивався до 1 року із затримкою, була помірна гіпотонія кінцівок. Голову почав тримати у 4 міс., сидіти — з 8 міс., пішов після 1 року, невпевнено, хитко. В 6 міс. обстежений у генетика у медико-генетичному центрі «ОХМАДИТ», спадкової патології не виявлено. За даними НСГ, у 1 р. 2 міс. виявлено ознаки порожнини Верге. У березні 2015 р. на фоні гострої респіраторно-вірусної інфекції з гіпертермією до 40 °С вперше в житті був епізод судом у вигляді клонічних скорочень верхніх кінцівок, тривалістю до 1 хв. Сімейний анамнез: у тітки за лінією батька був синдром Дауна, померла у віці 40 років.
При огляді соматичний статус дитини не порушений. На шкірі правого стегна пляма кольору кави з молоком 6 × 8 см (рис. 1). На шкірі сідниць 2 «монгольські» плями, невус на волосистій частині голови розміром 2 × 2 см (рис. 2). Окружність голови 56 см (+2 SD). Голова макроцефальної форми, чоло широке, підборіддя вузьке. Гіпертелоризм, епікант, «готичне» піднебіння. Гіпертрихоз шкіри плечей, вух. Зріст 109,5 см. Маса тіла 20 кг. Контакт із дитиною ускладнений. Не завжди реагує на власне ім’я. Виконує команди вибірково. Показує на прохання частини обличчя, кольори. Говорить близько 10 слів, мовлення нечітке. Поведінка гіперактивна, увага нестійка. Фотореакція жива, симетрична. Функції черепних нервів без патології. Хода не порушена. М’язові тонус та сила у кінцівках помірно знижені, D = S. Сухожилкові рефлекси живі, D = S. Черевні рефлекси живі. Розлади координації, патологічні рефлекси відсутні. Порушення функцій тазових органів відсутні.
/156.jpg)
Під час перебування у клініці проведено загальний і біохімічний аналізи крові, загальний аналіз сечі, патологічних відхилень не виявлено. За даними обстеження внутрішніх органів виявлено ознаки реактивних змін підшлункової залози, збільшення мезентеріальних лімфатичних вузлів. За даними УЗД щитоподібної залози, виявлено УЗ-ознаки дифузного зоба 2-го ст. Визначення маркерів патології щитоподібної залози (Т3, Т4, ТТГ) у крові показало зниження рівня тиреотропного гормона, наявність підвищення рівня антитіл до тиреопероксидази та тиреоглобуліну. Дитина консультована дитячим ендокринологом, встановлено діагноз: хронічний аутоімунний тиреоїдит, гіпертрофічна форма, еутиреоз.
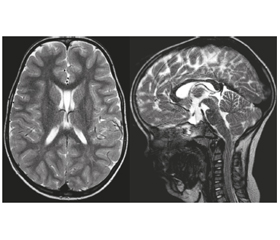
За даними ЕКГ виявлено синусовий ритм, ЧСС 112 уд/хв, порушення провідності по правій ніжці пучка Гіса. ЕЕГ без патологічних змін. МРТ головного мозку: МР-ознаки кісти прозорої перегородки (рис. 4). Висновок генетика: синдром церебрального гігантизму асоційований із дисплазією сполучної тканини. Висновок психіатра: наслідки органічного порушення і дисфункції головного мозку з порушенням психологічного розвитку, з недостатністю формування функції мовлення. Висновок психолога: первазивний розлад розвитку з помірною інтелектуальною недостатністю та порушенням формування мовлення. Висновок логопеда: загальне недорозвинення мовлення 1-го рівня. Висновок дерматолога: себорейний невус волосистої частини голови. Пігментний невус стегна правої кінцівки. «Монгольська» пляма сідниці. Висновок окуліста: міопія слабкого ступеня обох очей. Висновок ЛОР: хронічний аденотонзиліт.
/157.jpg)
Отже, беручи до уваги дані анамнезу (збільшений зріст; маса тіла; окружність голови при народженні; затримка розвитку статокінетичних навичок протягом першого року життя, що супроводжувалась м’язовою гіпотонією; особливості клінічного огляду та інструментального обстеження), можна встановити діагноз: синдром Сотоса із затримкою психомовленнєвого розвитку та РАС. Хронічний аутоімунний тиреоїдіт, гіпертрофічна форма, еутиреоз.
Висновки
Значною проблемою сучасної нейропедіатрії є збільшення частоти випадків РАС у дітей. Нерідко під маскою РАС може бути прихований інший патологічний стан, якому автоматично присвоюється діагноз «аутизм». З метою встановлення правильного діагнозу кожна дитина з підозрою на РАС повинна бути оглянута дитячим неврологом, за необхідності мають бути проведені додаткові лабораторні та інструментальні обстеження.
Синдром Сотоса є одним із патологічних станів із генетичною етіологією, в клінічній картині якого можуть спостерігатися порушення поведінки різного характеру та затримка психомовленнєвого розвитку, в тому числі симптомокомплекс РАС. Золотим стандартом діагностики цього захворювання є молекулярно-діагностичне дослідження з виявленням мутації гена NSD1. Однак за відсутності можливості провести такий аналіз діагноз може бути встановлений на підставі типової тріади симптомів: 1) макросомія (гігантизм); 2) специфічний фенотип (риси обличчя); 3) затримка когнітивного та мовленнєвого розвитку.
Перебіг синдрому Сотоса є непрогресуючим. Раннє лікування та психолого-педагогічна реабілітація дітей-пацієнтів дозволяє досягти покращення когнітивних, соціальних та мовленнєвих функцій.
Вітчизняні фахівці з дитячої психоневрології повинні бути мотивовані на пошук можливої етіології у кожної дитини з РАС. Адекватно встановлений діагноз дозволяє розпочати комплекс індивідуально розроблених лікувально-реабілітаційних заходів, який у багатьох випадках здатний суттєво поліпшити якість життя пацієнтів і рівень соціального функціонування, попередити їх інвалідизацію.
Список литературы
1. McCabe C., Claxton K., Tsuchiya A. Orphan drugs and the NHS: should we value rarity? // BMJ. — 2005 Oct 29. — Vol. 331(7523). — P. 1016-9.
2. Закон України «Про внесення змін до Основ законодавства України про охорону здоров’я щодо забезпечення профілактики та лікування рідкісних (орфанних) захворювань» № 1213-VII від 15 квітня 2014 року // Відомості Верховної Ради. — 2014. — № 26. — С. 894.
3. Puiu M., Dorica D. Rare diseases, from European resolutions and recommendations to actual measures and strategies // Maedica (Buchar). — 2010 Apr. — Vol. 5(2). — P. 128-131.
4. Stewart S., Peers K. Rare diseases: what do you need to know? // Nursing Times. — 2013. — Vol. 45. — P. 12-14.
5. Tatton-Brown K., Rahman N. Sotos Syndrome // European Journal of Human Genetics. — 2007. — Vol. 15. — P. 264-271.
6. Hyland S. Toward a behavioural phenotype for Sotos syndrome // Clin. Psy. D. Thesis. — University of Birmingham, 2011.
7. Sotos J., Dodge P. et al. Gigantism in childhood. A syndrome of excessively rapid growth and acromegalic features and a nonprogressive neurologic disorder // N. Engl. J. Med. — 1964 Jul 16. — Vol. 271. — P. 109-116.
8. Cole T.R.P., Hughes H.E. et al. Small cell lung carcinoma in a patient with Sotos syndrome: are genes at 3p21 involved in both conditions? // J. Med. Genet. — 1992. — Vol. 29. — P. 338-341.
9. Kurotaki N., Imaizumi K. et al. Haploinsufficiency of NSD1 causes Sotos syndrome // Nat. Genet. — 2002 Apr. — Vol. 30(4). — P. 365-6. Epub 2002 Mar 18.
10. Tatton-Brown K., Douglas J. Genotype-phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aber-rations // Am. J. Hum. Genet. — 2005b. — Vol. 77. — P. 193-204.
11. Sotos J.F. Overgrowth // Clin. Pediatr. — 1997. — Vol. 36. — P. 89-103.
12. Tatton-Brown K., Rahman N. Clinical features of NSD1-positive Sotos syndrome // Clin. Dysmorphol. — 2004. — Vol. 13. — P. 199-204.
13. Baujat G., Cormier-Daire V. Sotos syndrome Orphanet // J. Rare Dis. — 2007 Sep 7. — Vol. 2. — P. 36.
14. Sotos Syndrome: A Handbook for Families by Rebecca Rae Anderson, J.D., M.S., Bruce A. Buehler, M.D., G. Bradley Schaefer, M.D. (third edition, 2005).
15. Compton M., Celentana M. et al. A case of Sotos syndrome (cerebral gigantism) and psychosis // Psychopathology. — 2004 Jul-Aug. — Vol. 37(4). — P. 190-3. Epub 2004 Jul 2.
16. Sarimski K. Behavioral and emotional characteristics in children with Sotos syndrome and learning disabilities // Developmental Medicine and Child Neurology. — 2003. — Vol. 45. — P. 172-178.
17. Morrow J. et al. Autistic disorder in Sotos syndrome // Eur. J. Pediatr. — 1990. — Vol. 149. — P. 567-569.
18. Zapella M. Autistic features in children affected by cerebral gigantism // Brain Dysfunction 01/1970. — 1990. — Vol. 3. — P. 241-244.
19. Leventopoulos G., Kitsiou-Tzeli S. et al. A clinical study of Sotos syndrome patients with review of the literature // Pediatr. Neurol. — 2009. — Vol. 40. — P. 357-64.
20. Mouridsen S., Hansen M. Neuropsychiatric aspects of Sotos syndrome. A review and two case illustrations // Eur. Child Adolesc. Psychiatry. — 2002 Feb. — Vol. 11(1). — P. 43-8.
21. Finegan J., Cole T. Language and Behaviour in children with Sotos Syndrome // Journal of the American Academy of Child and Adolescent Psychiatry. — 1994. — Vol. 33(9). — P. 1307-15.
22. Hersh J., Cole T. et al. Risk of malignancy in Sotos syndrome // J. Pediatr. — 1992. — Vol. 120. — P. 572-4.
23. Tatton-Brown K., Rahman N. Clinical features of NSD1-positive Sotos syndrome // Clin. Dysmorphol. — 2004. — Vol. 13. — P. 199-204.
24. Wang X., Yeh S. et al. Identification and characterization of a novel androgen receptor coregulator ARA267-alpha in prostate cancer cells // J. Biol. Chem. — 2001. — Vol. 276. — P. 40417-40423.