
Международный неврологический журнал 2(2) 2005
Вернуться к номеру
Пероксисомные заболевания /Peroxisomal Disorders/
Авторы: Хьюго У. МОЗЕР, Энн Б. МОЗЕР, Medical University, Beterda, США
Рубрики: Неврология
Разделы: Справочник специалиста
Версия для печати
Пероксисомные нарушения — это группа генетически детерминированных заболеваний, понятых за последние 20 лет. Они подразделяются на две основные группы: 1. Нарушения пероксисомного биогенеза; 2. Недостаточности одиночных пероксисомных ферментов.
Нарушения пероксисомного биогенеза
В этой группе больных пероксисома нормально не образуется. Сейчас выявлено 13 таких нарушений. Их основным дефектом является аномалия в процессах, которые контролируют белки в пероксисоме. Это сложный процесс, он включает в себя более 30 отдельных генов, которые сейчас называются пероксинами (РЕХ).
Сейчас показано, что дефекты в 13 из этих РЕХ генов связаны с болезненными состояниями человека. Эти нарушения даны в табл. 1.
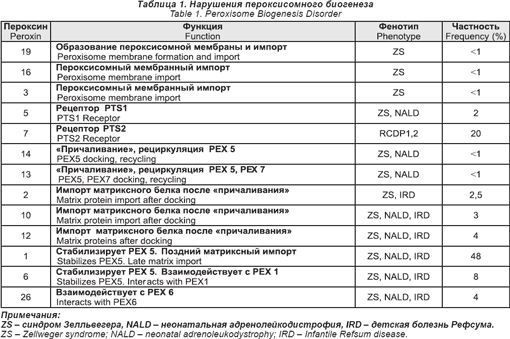
Клинические фенотипы этих нарушений подразделяются на две основные категории:
1) Спектр болезней Зелльвегера (ZS), неонатальный ALD (NALD), детской болезни Рефсума (IRD). Эти нарушения были описаны и названы задолго до того, как стали известны их основная природа и связь с пероксисомой. Сейчас считается, что они образуют клинический спектр, который изменяется в отношении тяжести заболевания, где ZS — самая тяжелая, а IRD — самая легкая (хотя его все еще связывают с большими дефицитами), а NALD — промежуточная. Больные с ZS получают тяжелые поражения при рождении и имеют характерные черты лица, тяжелую гипотонию, трудности при кормлении, глубокую задержку психомоторного развития, пигментарную дегенерацию сетчатки глаза, потерю слуха, увеличенную печень, многие умирают до двух летнего возраста. У больных с IRD более легкие аномалии, психомоторная дегенерация различной степени, пигментарная дегенерация и нарушения слуха. Большую пользу может принести им специальное общение, физическая и трудовая терапия, слуховые аппараты. Они могут дожить до взрослого возраста. Больные с NALD напоминают больных с IRD, за тем исключением, что у них может развиться прогрессивная лейкодистрофия. Чаще всего проявляется в возрасте от 2-х до 6-ти лет, но у одного больного она появилась только на 40-м году жизни.
В табл. 1 показано, что 12 из дефектов PЕХ могут быть связаны с каждым их трех фенотипов (ZS, NALD, IRD). Пока полного понимания этого еще нет, проведенные исследования выращенных фибробластов показали, что клеточные линии от больных с более легкими фенотипами сохранили некоторое их количество, чтобы импортировать пероксисомный белок.
2) Дефекты PЕХ 7 связаны с клиническим фенотипом ризомелических хондродисплазийных пятен (RCDP). При полной экспрессии — это тяжелое заболевание, которое включает укорачивание конечностей, глубокую задержку психомоторного развития, катаракту, характерные черты лица, ихтиоз и сильную зернистость эпифизов и корональные щели тел позвонков на рентгенограмме. Большинство детей умирают в течение первых пяти лет.
Недавно было показано, что у некоторых больных с дефицитом РЕХ 7 синдром намного слабее. У этих больных мутации слабее, и сохраняются некоторые функции РЕХ 7. Некоторые их этих больных имеют катаракты без укорачивания конечностей. Клинические признаки других похожи на таковые болезни Рефсума с развитием периферической невропатии и пигментарной дегенерации сетчатки глаз в подростковом возрасте или ранней юности.
Все эти нарушения пероксисомного биогенеза имеют аутосомный рецессивный способ наследования. Их общая заболеваемость, по оценкам, составляет 1: 50 000. Их можно диагностировать при помощи серии биохимических анализов плазмы крови и эритроцитов (см. табл. 2). Перинатальный диагноз возможен для всех. Для определения молекулярных дефектов необходимы специальные анализы. Стейнберг и его сотрудники недавно разработали алгоритм, позволяющий определить большинство из них.
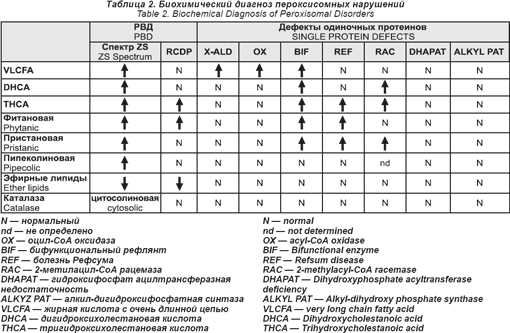
Специальной терапии пероксисомных нарушений нет. Некоторым легко больным очень помогла мультидисциплинарная поддерживающая и реабилитационная терапия.
Нарушения одиночного пероксисомного белка
Эти нарушения даны в таб. 3
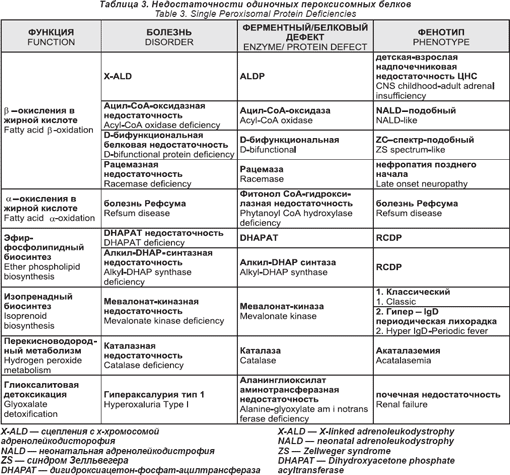
Сцепленная с X-хромосомой адренолейкодистрофия
Сцепленная с X-хромосомой адренолейкодистрофия — самое распространенное пероксисомное заболевание. По всему миру заболеваемость его составляет 1:17 000. Дефектный ген, который сейчас называется ABCD1, был картирован на Xq28. Он является геном надсемейства белков АВС транспорта. Он кодирует пероксисомный белок или мембраны, ALDP. Скопление насыщенных жирных кислот с очень длинными цепями (VLCFA), в основном гексакосаноиновой кислоты (С26:0), является основной биохимической аномалией, а определение избытка VLCFA в плазме — наиболее часто применяемый диагностический анализ.
Х-ALD имеет 3 основных фенотипа:
1) Церебральный детства. Чаще всего проявляется между 4-м и 8-м годами жизни. Первичные симптомы напоминают гиперактивный дефицит внимания, но через год-два прогрессируют с более тяжелыми расстройствами поведения, визуомоторными и слуховыми дефицитами, деменцией, а позже — прогрессирующими трудностями в координации, речи, глотании, спастичностью и слабостью.
2) Адреномиелоневропатия (AMN). AMN представлена у молодых людей как медленно прогрессирующий парапарез, нарушенное чувство вибрации и положения и нарушения функции сфинктера. Она прогрессирует медленно, на это уходят десятки лет. В большинстве случаев интеллект сохраняется, но примерно у 20% больных также развивается церебральное поражение, когда это случается, болезнь прогрессирует быстрее. Приблизительно у 50% женщин, гетерозиготных к Х-ALD, AMN развивается в среднем возрасте или позже, но она обычно легче, чем у мужчин.
3) Фенотип «Аддисон только»: эти больные страдают первичной адренокортикальной недостаточностью, их нельзя отличить от больных с болезнью Аддисона, за тем исключением, что уровни VLCFA в плазме крови у них возрастают. Чаще всего этот фенотип проявляется в детстве. У большинства Х-ALD больных с фенотипом «Аддисон только» также в дальнейшем развивалось церебральное поражение или AMN. У женщин, гетерозиготных к Х-ALD, недостаточность надпочечников встречается редко.
Диагноз
У мужчин Х-ALD надежно диагностируется анализом VLCFA в плазме. Результаты теста не соответствуют норме уже в день рождения. Однако ложноотрицательные результаты встречаются примерно у 20% женщий, обязательно имеющих гетерозиготы к Х-ALD. Анализ мужчин точно определяет гетерозиготы. Возможен пренатальный диагноз, когда проводится анализ VLCFA или анализ мутации.
МРТ выявляет характерное нарушение у большинства мальчиков с Х-ALD. Наиболее тяжелым это нарушение является в теменно-затылочной областях. Могут также иметь место необычные формы: поражение лобной доли или односторонние поражения. У больных с AMN МРТ мозга обычно показывает норму. В то время как МРТ изменения в церебральной форме часто имеют довольно характерные особенности, диагноз должен быть подтвержден биохимическим анализом или анализом мутации.
Лечение
Если функция надпочечников нарушена, то у всех больных с Х-ALD необходимо проверить функцию надпочечников и назначить им стероидную заместительную терапию надпочечников. Это может спасти им жизнь. Как кажется, гормональное замещение в надпочечниках не действует на неврологическую прогрессию. Было показано, что трансплантация кроветворных клеток полезна для мальчиков с церебральным ALD, но только в том случае, когда она еще легкая (IQ выше 80, а баллов по шкале МРT Лeса меньше 9). Это важная причина, по которой диагностировать Х-ALD следует рано, до наступления тяжелого поражения.
Профилактика
Рекомендуется, чтобы все родственники больных с Х-ALD, которые подвергаются риску заболеть, прошли анализ на VLCFA в плазме крови. Это позволит выявить женщин-носительниц Х-ALD. Они должны получить генетические рекомендации. Этот скрининг позволит выявить бессимптомных или слабосимптомных мальчиков в то время года, когда трансплантация все еще может принести пользу. Пренатальный диагноз Х-ALD точен.
Болезнь Рефсума
Только недавно было продемонстрировано, что болезнь Рефсума — пероксисомное нарушение. Оно диагностируется по анализу уровней фитановой кислоты, ферментному анализу или анализу мутаций. Эффективны терапия детей с ограниченным количеством фитановой кислоты и плазматический обмен.
D-Бифункциональная энзимопатия
D-бифункциональная энзимопатия — заболевание часто встречающееся. Клинически оно напоминает адренолейкодистрофию Зелльвегера или неонатальную. Удивительно, что 15% больных с этими фенотипами болеют скорее D-бифункциональной энзимопатией, чем нарушением пероксисомного биогенеза.
1. Weller S., Gould S.J., Valle D. Peroxisome biogenesis disorders. Annu Rev Genomics Hum Genet 2003;4:165-211.
2. Gould S.J., Raymond G.V., Valle D. The Peroxisome Biogenesis Disorders. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., eds. The Metabolic and Molecular Bases of Inherited Diseases. Eighth Edition ed. New York: McGraw Hill, 2001:3181-3217.
3. Braverman N., Chen L., Lin P. et al. Mutation analysis of PEX7 in 60 probands with rhizomelic chondrodysplasia punctata and functional correlations of genotype with phenotype. Hum Mutat 2002;20:284-297.
4. Steinberg S., Chen L., Wei L. et al. The PEX gene screen: Molecular diagnosis of peroxisome biogenesis disorders in the Zellweger syndrome spectrum. Molecular Genetics and Metabolism 2004; in press.
5. Moser H.W., Smith K.D., Watkins P.A., Powers J., Moser A.B. X-linked adrenoleukodystrophy. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. Eighth Edition ed. New York: McGraw Hill, 2001:3257-3301.
6. Moser A.B., Kreiter N., Bezman L. et al. Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls. AnnNeurol 1999;45:100-110.
7. Bezman L., Moser A.B., Raymond G.V. et al. Adrenoleukodystrophy. incidence, new mutation rate, and results of extended family screening. AnnNeurol 2001;49:512-517.